New UNC School of Medicine research led by Martina Gentzsch, PhD, and Carla Ribeiro, PhD, suggests that cystic fibrosis drugs used to correct the faulty CFTR protein would work better when the patients’ airways are inflamed.

Media contact: Mark Derewicz, 984-974-1915, mark.derewicz@unchealth.unc.edu
CHAPEL HILL, NC – A new UNC School of Medicine study shows that two cystic fibrosis (CF) drugs aimed at correcting the defected CFTR protein seem to be more effective when a patient’s airway is inflamed. This is the first study to evaluate the efficacy of these drugs under inflammatory conditions relevant to CF airways.
This work, published in the European Respiratory Journal, offers a greater understanding of the efficacy of the CF drugs Lumacaftor and Ivacaftor and provides a new model for pre-clinical evaluation of CFTR-modulating therapeutics that scientists in academia and in the pharmaceutical industry are now pursuing.
“Our finding is counterintuitive,” said first author Martina Gentzsch, PhD, associate professor of cell biology and physiology. “We were surprised that the effectiveness of CFTR modulators might depend on the state of the airway, inflamed or not.”
CF, one of most common genetic diseases, occurs when a person acquires two defective versions of the CFTR gene from their parents. The result is an airway unable to stay hydrated, which is critical for moving naturally occurring mucus out of the respiratory system. This results in airway obstruction, chronic infections, and inflammation. Therapies, such as hypertonic saline, have helped limit these symptoms while substantially extending the lives of CF patients. Yet, average life expectancy is still just 37 years.
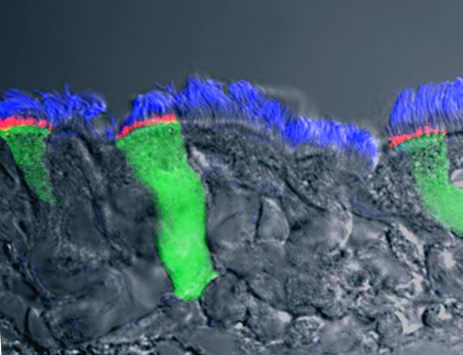
In the past decade, scientists created new therapies, such as lumacaftor and ivacaftor, to correct the CFTR protein. But these drugs, used in combination, have not been highly beneficial for most CF patients. Gentzsch, senior author Carla Ribeiro, PhD, and UNC colleagues designed lab experiments to find out why. They exposed primary cultures of CF human bronchial airway cells to the infectious and inflammatory milieu found in the airways of CF patients. To their surprise, they found lumacaftor and ivacaftor were more effective in correcting the CFTR defect when the CF cultures were inflamed.
“We found that under inflammatory conditions, there was more functional CFTR available for hydrating the CF airways,” said Ribeiro, associate professor of medicine and cell biology and physiology. “Because CF patients are also under anti-inflammatory therapy, researchers and doctors need to consider the interference of anti-inflammatory drugs with CFTR modulators in future studies.”
Gentzsch and Ribeiro said that as more CFTR modulator combination therapies become available, it will be critical to evaluate pre-clinical efficacy under conditions that mimic inflamed CF airway epithelia.
“We are convinced that these sorts of studies will be necessary in order to optimize current and future CF therapies,” Ribeiro said.
Gentzsch and Ribeiro are members of the UNC Marciso Lung Institute and the UNC Cystic Fibrosis Research Center. Other authors include Deborah Cholon, PhD, Nancy Quinney, Susan Boyle, and Mary Martino, all at the UNC Marsico Lung Institute and the UNC Cystic Fibrosis Research Center. The Cystic Fibrosis Foundation and Cystic Fibrosis Research, Inc. funded their study.