Giving mucus-thinning therapies in early childhood might delay the worst symptoms and infections associated with CF, according to a study led by Charles Esther, MD, PhD, and Marianne Muhlebach, MD. Their paper is the cover story of Science Translational Medicine.
Giving mucus-thinning therapies in early childhood might delay the worst symptoms and infections associated with CF, according to a study led by Charles Esther, MD, PhD, and Marianne Muhlebach, MD. Their paper is the cover story of Science Translational Medicine.
Media contact: Mark Derewicz, 984-974-1915, mark.derewicz@unchealth.unc.edu
April 3, 2019
CHAPEL HILL, NC – The build-up of abnormally thick mucus and the associated inflammation appear to be the initiating cause of damage to the lungs of children with cystic fibrosis (CF), rather than bacterial infections, according to a UNC School of Medicine study published in Science Translational Medicine.
The research suggests that doctors might be able to delay the onset of lung disease in young children born with CF, and possibly allow them to live significantly longer. The key would be to use early treatments that thin out mucus.
“How lung disease starts in the youngest kids with CF hasn’t been well understood, but with this study we can see what processes come first and develop strategies to target them to help patients,” said co-first author Charles R. Esther, Jr., MD, PhD, professor of pediatrics at the UNC School of Medicine and member of the UNC Marsico Lung Institute.
More than 30,000 people in the United States have CF, a genetic disorder that leads to the production of abnormally thick mucus in the lungs and windpipe. Children with CF are born with mostly normal lung functions, but eventually develop lung inflammation, recurrent bacterial pneumonia, and progressive scar-like changes to lung tissue. Life expectancy for CF patients is less than 40 years.
Understanding the first stages of the CF disease process in the lungs would, in principle, allow researchers to develop early treatments to delay or prevent that disease process. But studying early CF has been very challenging, in part because animal models of CF do a poor job of mimicking the human disease.
Esther, co-first author Marianne Muhlebach, MD, professor of pediatrics at the UNC School of Medicine, and a multidisciplinary group of researchers at UNC-Chapel Hill – collaborating with Australian researchers led by Dr. Stephen Stick – looked not at animal models but at CF patients themselves. The researchers analyzed the contents of “lavage” fluid that had been used to rinse the lungs of 46 young children with CF as part of an Australian clinical study, and compared the samples to lavage fluid from 16 other Australian children with asthma and other non-CF airway ailments.
Bacterial infection has been suspected as an early driver of lung damage in CF, but the researchers found little evidence of bacteria in the young CF patients’ lavage fluids. In fact, they found more bacteria on average in the non-CF samples.
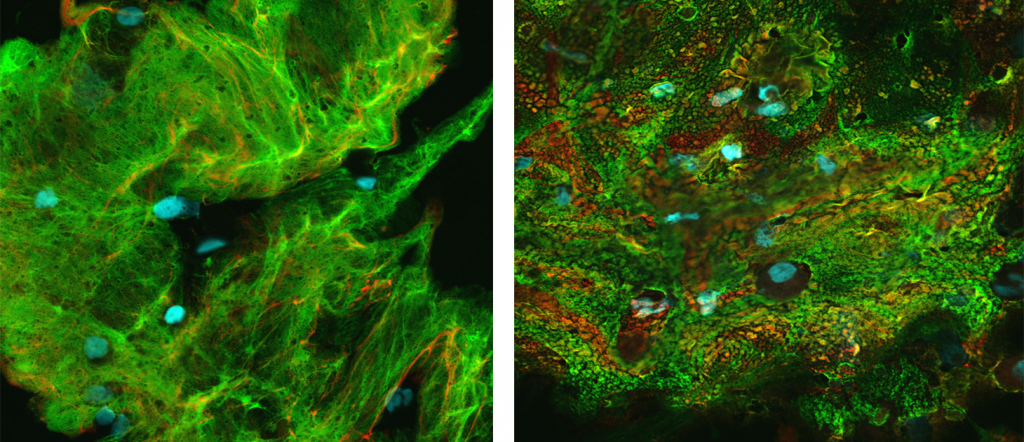
Here’s the key difference: the CF samples contained more evidence of mucus, implying that CF causes patients to produce or accumulate more of it. Also, the CF mucus was much more likely to be a more solid “flake” form with a high concentration of proteins called mucins.
And here’s another important finding: areas of the CF lungs where serious damage had not yet occurred still featured an abnormally high concentration of mucins and signs of inflammation, even without strong evidence of infection.
Esther, Muhlebach, and colleagues now hypothesize that in young CF patients, mucus that is secreted – as it should be during ordinary viral respiratory infections – isn’t fully cleared because it is abnormally thick. It builds up in the airway and creates a low-oxygen condition in airway-lining cells. This triggers inflammation, which stimulates more mucus secretion and more inflammation.
This “positive feedback loop” ultimately results in scarring and progressive loss of lung function. Recurrent infections with dangerous bacteria begin at some point during this process and worsen the disease course. But those infections, the researchers say, are likely not the earliest drivers of lung damage.
The finding that lung inflammation in CF children appears to result initially from thick airway mucus, not bacterial infection, suggests that early mucus-thinning interventions might delay the course of the disease.
The study investigators tested several prospective mucus-thinning compounds on the mucin flakes isolated from the CF lavage samples. They found that two FDA-approved drugs, DNase and N-acetylcysteine, which are used as mucus-thinners in CF patients, did not work well in dissolving the flakes. A third compound, dithiothreitol, did work well but is too toxic for human use. An experimental compound called P2062, developed by UNC researchers in collaboration with biotech company Parion Sciences, also appeared to work well at reducing mucin flakes though it has not yet been tested in people.
“We are now trying to find new therapeutic agents that could help remove these flakes in CF patients,” Esther said.
Other authors of the Science Translational Medicine paper were Camille Ehre, PhD, David B. Hill, PhD, Matthew C. Wolfgang, PhD, Mehmet Kesimer, PhD, Kathryn A. Ramsey, PhD, Matthew R. Markovetz, PhD, Ian C. Garbarine, M. Gregory Forest, PhD, Ian Seim, PhD, Bryan Zorn, Cameron B. Morrison, Martial F. Delion, PhD, and Richard C. Boucher, MD, all at UNC-Chapel Hill at the time of the study; William R. Thelin, PhD, and Diane Villalon PhD, of Parion Sciences in Durham, NC; Juan R. Sabater, MD, of Mount Sinai Medical Center in Miami Beach, FL; Lidija Turkovic, PhD, and Stephen M. Stick, MD, of the University of Western Australia; and Sarath Ranganathan, MD, PhD, of the University of Melbourne.
The research was funded by grants from the U.S. National Heart, Lung and Blood Institute, National Institute of Environmental Health Sciences, National Institute of General Medical Sciences, Australia’s National Health and Medical Research Council, and the Cystic Fibrosis Foundation.