Researchers at the UNC School of Medicine led by Justin Wolter, PhD, and colleagues at the UNC Eshelman School of Pharmacy and Queen Mary University of London, teamed up to publish a comprehensive analysis of what goes awry in brain cells lacking the sacsin protein.

CHAPEL HILL, NC – Scientists at the UNC School of Medicine and UNC Eshelman School of Pharmacy, in collaboration with a team from Queen Mary University of London, have illuminated the molecular events underlying an inherited movement and neurodegenerative disorder known as ARSACS – Autosomal recessive spastic ataxia of Charlevoix-Saguenay, named for two Quebec valleys where the first cases were found.
Children with ARSACS typically display difficulties with walking in the second year of life and an expanding array of neurological problems thereafter. In the cerebellum – an area of the brain which coordinates movement and balance – neurons called Purkinje cells die in individuals with ARSACS. Most patients are wheelchair-bound by their 30s-40s, and have a shortened lifespan averaging in the mid-50s.
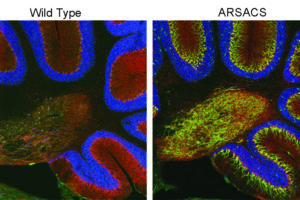
The disorder is caused by the mutation and functional loss of a gene called SACS that encodes a very large protein called sacsin, which has been hard to study directly in part because of its unwieldy size. Relatively little has been known about its normal functions, and how its absence leads to disease. But in a study published in Cell Reports, the collaborating researchers performed the most comprehensive analysis of what happens in cells when sacsin is missing.
“We tried to take an unbiased approach to understand what goes wrong when cells lose sacsin. Our results suggest that the death of Purkinje cells in ARSACS may possibly result from changes in neuronal connectivity and synaptic structure,” said study co-senior author Justin Wolter, PhD, a postdoctoral researcher at the UNC Neuroscience Center.
The other co-senior author of the study was Paul Chapple, PhD, a professor of molecular cell biology at Queen Mary University of London.
The study began with Chapple lab and the UNC-Chapel Hill team working without knowledge of the other. “This project was started by Tammy Havener in the UNC Eshelman School of Pharmacy, then three postdoctoral researchers from different UNC departments jumped on board – Wen Aw, Katherine Hixson, and myself,” Wolter said. “When we realized that Lisa Romano in the Chapple lab had made similar discoveries using different approaches we all decided to join forces and move forward together. I think it’s a beautiful example of how open science and collaboration pays off for the community.”

For this study, the researchers used several -omics based techniques in cultured human cells to examine how the loss of sacsin changes protein levels and cellular organization. They confirmed the presence of defects that had been noted in prior studies, such as the abnormal aggregation of filament-forming structural proteins, and defects in the numbers and dynamics of mitochondria, both of which are frequently observed in many neurodegenerative diseases.
But they also found many abnormalities that hadn’t been identified before. These included the overabundance of a protein called tau and altered dynamics of microtubules, which are intracellular transport tracks regulated by tau. The researchers found that the consequence of this change in trafficking was that many proteins did not get to the proper location in the cell. Particularly affected were “synaptic adhesion” proteins, which help neurons form and maintain synapses – connections neurons use to send signals to each other. In line with these observations, the team found changes in synaptic structure in the ARSACS mouse model. Importantly, these changes occur before the onset of neurodegeneration.
These discoveries expand the picture of how sacsin regulates multiple cellular processes. They also suggest the possibility that Purkinje cells – the neurons that seem most affected in ARSACS – might die because they lack connections with other neurons. The researchers will follow up with more in-depth studies of these changes in the brain to understand whether or not this neurodegenerative disease is rooted in processes that unfold during brain development.
Although ARSACS affects probably only a few thousand individuals worldwide, this kind of research could have much broader implications, the researchers noted.
“There appear to be multiple overlaps between ARSACS and other brain disorders,” Chapple said. “We showed for example that there’s disruption of tau biology in cells lacking sacsin, and of course abnormalities in tau are also a well-known feature of Alzheimer’s disease. So we think studying this rare neurological condition could provide insights into much more common ones.”
“Much work remains to be done to understand the mechanisms by which synaptic connectivity is affected and whether it is contributing to neuronal death,” Wolter said. “But, if it is, it could inform future therapeutic approaches.”
The Cell Reports paper titled “Multi-omic profiling reveals the ataxia protein sacsin is required for integrin trafficking and synaptic organization” was co-authored by Lisa Romano, Wen Yih Aw, Kathryn Hixson, Tatiana Novoselova, Tammy Havener, Stefanie Howell, Charlotte Hall, Lei Xing, Josh Beri, Suran Nethisinghe, Laura Perna, Abubakar Hatimy, Ginevra Chioccioli Altadonna, Lee Graves, Laura Herring, Anthony Hickey, Konstantinos Thalassinos, J. Paul Chapple and Justin Wolter.
Funding was provided by Fondation de l’Ataxie Charlevoix-Saguenay, the UK Biotechnology and Biological Sciences Research Council, Ataxia UK, and the National Institutes of Health (T32HD040127, R01 GM138520).
Media contact: Mark Derewicz, UNC School of Medicine, 919-923-0959