Jane Little, MD, and Maria Boucher, MD, both in the UNC School of Medicine, detail the current struggles of patients with sickle cell disease and how we can help.
CHAPEL HILL, N.C. – Since it’s discovery in 1910, sickle cell disease has been considered a death sentence for those that inherited it. But over the years, dedicated pediatric programs and research initiatives have greatly improved patient care and life expectancy.
Giving these patients the care they require still presents a number of challenges, including inadequate funding for sickle cell programs, lack of research, and limited access to healthcare.
UNC School of Medicine’s Jane Little, MD, director of the UNC Comprehensive Sickle Cell Disease program, and Maria Boucher, MD, a pediatric hematologist-oncologist and director of the Pediatric Sickle Cell program, detail the current struggles of patients with sickle cell disease and how we can help.
What is Sickle Cell Disease?
Sickle cell disease, also known as sickle cell anemia, is an inherited red blood cell disorder that is woefully underfunded and under researched. The disease affects about 5,000 North Carolinians, predominantly African Americans and Hispanics.
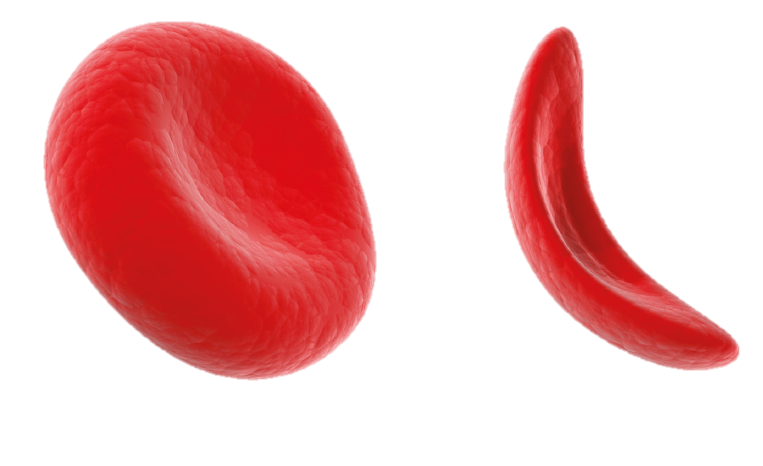
Normally, red blood cells look like throwing discs. In patients with sickle cell disease, affected blood cells resemble the curved blade of a sickle. As these abnormally shaped red blood cells travel through the blood vessels, they cause traffic jams and block blood supply to organs and other areas of the body. The subsequent blockage can then cause inflammation, debilitating pain, fatigue, and many other health problems.
“People with sickle cell are superheroes,” said Little, a UNC Health hematologist who specializes in the care of sickle cell patients. “They’re managing hemoglobin levels that would make me swoon. They have to put up with fatigue that would stun the rest of us. But they’re always on the edge. And any little thing that triggers their sickle cell disease can be life-threatening.”
Medicine, Research, and the Lack Thereof
There have been a few medical advancements in the past several years to help prevent or reduce the production of sickle-shaped red blood cells.

In 2017, the FDA approved several treatments for sickle cell disease, including L-glutamine, crizanlizumab-tmca, and voxelotor. Some medications can run upwards of $100,000 a year. For families with more than one member with sickle cell disease, even the more affordable medications can be a significant financial burden.
Sickle cell disease is chronically under-funded and receives little awareness, largely due to systemic racial disparities, according to Little. Patients with other hereditary chronic illnesses, such as cystic fibrosis (CF) and hemophilia, are typically Caucasians and have access to pharmaceutical funding, sustained resources, and support registries, which have not been accessible to patients with sickle cell disease.
Of the scant research being conducted on sickle cell disease, much is concentrated on curative treatments like gene therapy and stem cell therapy. These therapies, although promising and ingenious, often involve long hospital stays and incur harsh side effects that may hinder young patient’s social lives and quality of life, according to Little and Boucher.
“There’s a lot of stuff we don’t know about sickle cell disease because we don’t have 50 years of registries informing us about patient care and quality,” said Little. “Not everyone with sickle cell is going to qualify for gene therapy or the stem cell cures. It’s like, we haven’t built a dock and the boat that’s coming for our patients is tiny.”
A Tough Transition
Patients are under medical care for their entire lifetimes. With more people living longer with sickle cell disease than ever before, patients will need to transition from well-established pediatric sickle cell programs to adult programs. The latter of which are sparse and lack the expertise and financial safety nets.

The transition for pediatric to adult programs can be particularly risky for young adult patients because they lose their long-standing connection with their pediatric providers. Lapses in medications, forging new patient-provider relationships, and lack of healthcare access can be incredibly stressful for these patients.
With many sickle cell programs lacking specific transition programs, providers do their best to communicate with one another and prepare patients for their transition well ahead of time.
“The more we do to try either to treat or prevent sickle cell disease while they are kids, the better off they’ll go to the adult world,” said Boucher. “We work really closely with the adult programs to have this transition process go as well as it can, but the more resources we have and the more we do upfront, the better.”
Because North Carolina is a large state, it can be challenging for patients to quickly access the care they require; the major sickle cell programs are in the Triangle region.
For example, patients in UNC’s Comprehensive Sickle Cell Disease program typically travel 1.5 hours for their medical care on average. To alleviate the issues with healthcare access, many providers are travelling to satellite clinics during the work week or attending community outreach events.
“We need to go to the patient instead the other way around,” said Boucher. “Because North Carolina is such a huge state, I had a child come for a special transfusion who had to drive three hours one way to get to me.”
How to Help
Sickle cell disease providers, like Boucher and Little, rely heavily on funding from the Governor’s NC Appointed Council on Sickle Cell Disease and Other Blood Disorders and other state funding for outreach and longitudinal care. But more is needed to help their patients thrive.

On September 23, the UNC Comprehensive Sickle Cell Program is hosting a large-scale fundraiser to support Pediatric to Adult Transition programs in our North Carolina clinics and the Malawi Sickle Cell Clinic. The Full Court Press for Sickle Cell Basketball Tournament Fundraiser will be providing food, a blood drive, a bone marrow drive, and a knock-out basketball tournament on the UNC-Chapel Hill practice court.
Patients with sickle cell disease need blood transfusions and bone marrow transplants fairly often. African Americans are much more likely to be a blood or bone marrow match for someone with sickle cell disease, however, African Americans are less likely to donate blood or sign up for the bone marrow registry, making it much harder for sickle cell patients to have a match and get the care that they need.
“We are all part of the sickle cell community, not just the family members,” said Little. “These are North Carolinians, our neighbors. These are children and young adults, who are suffering more than you know and we should support them. Anything we can do to support these young people who are struggling, I’m all for it!”
Media contact: Kendall Daniels, Communications Specialist, UNC Health | UNC School of Medicine